Metabolic acidosis is generally defined by the presence of a low serum bicarbonate concentration (normal range 22-28 mEq/L), although occasionally states can exist where the serum bicarbonate is normal with an elevated anion gap (e.g., patients with a lactic acidosis who have received a bicarbonate infusion or patients on hemodialysis). In general, a metabolic acidosis is associated with a low urine pH but depending on the presence or absence of a respiratory alkalosis, this may also be normal or elevated. Thus, a patient can have an acidosis but not be acidemic.
Metabolic acidoses occur when there is excess acid in the plasma. In the basal state, the body generates about 12,000 to 13,000 mmol of carbon dioxide (CO2), and 1-1.5 mmol per kilogram body weight of nonvolatile acid. The body has a large buffering capacity, with CO2-HCO3 as the major buffer system. The two major routes of acid excretion are the lungs (for CO2) and the kidneys (for nonvolatile acids)
A metabolic acidosis can be caused by three major mechanisms: 1) increased acid production; 2) bicarbonate loss; and 3) decreased renal acid excretion
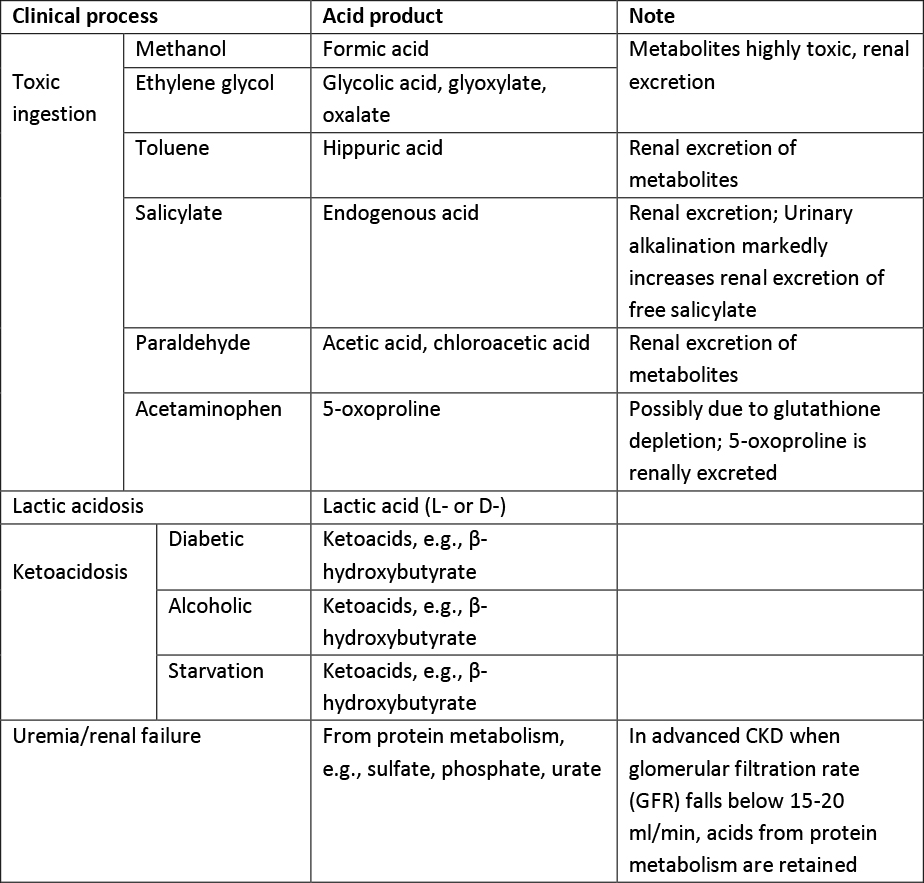
Increased acid production leads to anion-gap (AG) metabolic acidosis, and involves a variety of different clinical processes, see Table 1. An anion gap acidosis may also result for ingestion of an acid load.
Both bicarbonate loss and decreased renal acid excretion lead to normal-anion gap (NG) metabolic acidosis. When there is HCO3 loss, chloride is retained to maintain electrical neutrality. The different clinical processes are summarized in Table 2.
Toxic ingestions are common causes of AG metabolic acidosis. The commonest causes are methanol and ethylene glycol intoxication. These alcohols are quickly absorbed from the GI tract. Peak serum levels are usually reached within 1-2 hours. Immediately following ingestion, there is a large serum osmolar gap due to the presence of unmeasured, small, non-charge molecules. However, as these parent alcohols go through a two-step metabolism (via alcohol dehydrogenase and aldehyde dehydrogenase), the osmolar gap resolves while an anion gap acidosis develops. The toxicities mainly come from their metabolites.
Formic acid is the final metabolite of methanol, and glycolate, glyoxylate and oxalate are end metabolites of ethylene glycol. They accumulate and cause end-organ damage once the parent alcohol reaches a critical serum level (~20 mg/dl). However, because of slow hepatic metabolism, there is usually a latent period of 24-48 hours before these toxicities manifest, especially if there is co-ingestion of alcohol (which competitively inhibits alcohol dehydrogenase). If treatment is not instituted, permanent damage may ensue. Both methanol and ethylene glycol are excreted primarily by the kidneys, though the lung contributes to some degree of methanol elimination.
Salicylates can also cause AG metabolic acidosis. Salicylates are readily absorbed from the small intestine, and are metabolized in the liver through glycine conjugation. The amount of drug excreted unchanged in the urine is small, but can be dramatically increased with alkalization of urine due to trapping in the proximal tubule in a high pH environment preventing reabsorption. A similar mechanism prevents movement of salicylates across the blood-brain barrier in the setting of an alkalosis reducing the neurotoxicity associated with this drug. This is important because many patients with salicylate ingestion have a respiratory alkalosis due to stimulation of the respiratory center and this should not be reversed as it is protective. Similarly, carbonic anhydrase inhibitors, while they will increase urinary excretion of salicylic acid, are contraindicated because they lower extracellular pH and promote entry of the drug into the brain. The salicylic acid itself is not thought to be a significant contributor to the metabolic acidosis. The mechanism for the AG metabolic acidosis in salicylate overdose is still unclear, but is thought to be secondary to inhibition of the Krebs cycle and subsequent accumulation of organic acids, e.g., lactic acid and ketoacids.
Inhalation of toluene can lead to both AG and NG metabolic acidosis. The AG is caused by the metabolite of toluene, hippuric acid. However, hippuric acid is rapidly excreted in the urine and as the anion is exceted, the anion gap falls with a persistent NG acidosis. In general, these anions are excreted with potassium and as a result, toluene ingestion can lead to marked hypokalemia.
5-oxoproline is an increasingly recognized cause of an AG metabolic acidosis. It results from depletion of gluthathione. This prevents feedback inhibition of the gamma-glutamyl cycle and accumulation of oxoproline. It leads to an anion gap metabolic acidosis but may also cause a NG acidosis and hypokalemia due to rapid renal excretion of the anion. It is usually seen in patients with chronic, high dose acetaminophen intake and particularly effects older women. Other risk factors include sepsis, liver and/or kidney dysfunction.
L-Lactic acidosis is caused by either lactic acid overproduction from tissue hypoxia (Type A lactic acidosis) or lactic acid underutilization from thiamine deficiency/liver diseases or inihibition of oxidate phosphorylation, usually by a drug (type B lactic acidosis). Type A lactic acidosis is the most commonly seen in clinical practice and typically patients are hypotensive with obvious poor tissue perfusion. Drugs associated with a Type B lactic acidosis include metformin, phenformin, nucleoside reverse transcriptase inhibitors and propofol.
D-lactate is unique, as it is not metabolized by L-lactate dehydrogenase in human. It occurs in patients with short-bowel syndrome. When the small bowel is bypassed, large amount of carbohydrates are delivered to the colon, where there is an abundance of gram-positive anaerobes (e.g., Lactobacilli). Carbohydrates are metabolized into D-lactate which are then absorbed. Since D-lactate is not measured routinely when serum lactate is ordered, it should be specifically requested if D-lactic acidosis is suspected
Ketoacidosis occurs when there is an increased conversion of fatty acids to ketoacids (acetoacetate, β-hydroxybutyrate) in several pathological conditions, especially when insulin is lacking. In diabetic ketoacidosis, a NG metabolic acidosis is often encountered later in the course due to renal excretion of ketoacids. This also explains the severe total body potassium deficit usually seen in these patients.
Chronic kidney disease with decreased renal function is a common cause of metabolic acidosis. In the early phases with moderate functional decline (stage 3 & early stage 4), the kidney is still able to excrete organic acids, therefore AG acidosis is not common. Patients usually manifest a NG metabolic acidosis due to decreased ammonium excretion. Once renal function declines to a critical level, usually at late stage 4, acids from protein metabolism are retained, resulting in an AG metabolic acidosis.
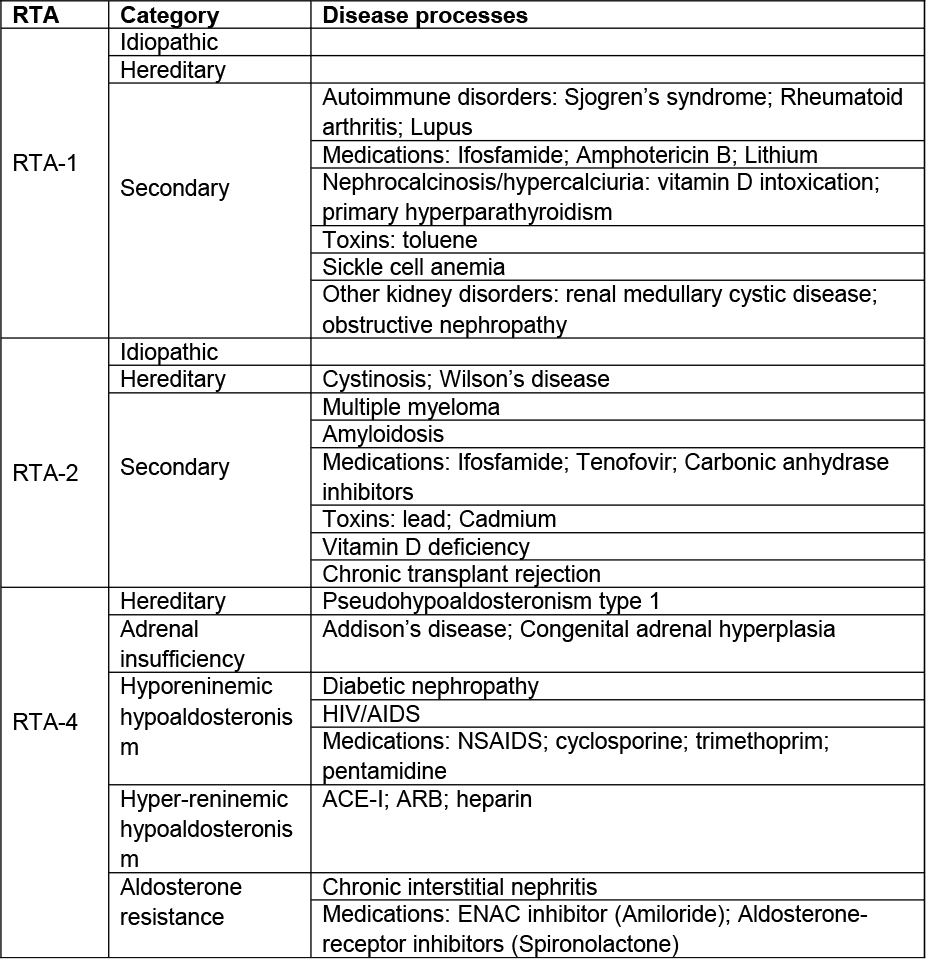
There are three major types of renal tubular acidosis (RTA): 1) type 1 (distal, RTA-1); 2) type 2 (proximal, RTA-2) and 3) type 4 (distal from low aldosterone or aldosterone resistance, RTA-4). RTA-1 results from a defect in distal tubular acid excretion as a result of decreased H+ secretion or back leak of secreted hydrogen. It can be severe and results in progressive HCO3 loss (serum concentration <10 meq/L). Urine pH in these patients is typically above 5.5 despite the metabolic acidosis. It stimulates bone resorption, and results in hypercalciuria and nephrocalcinosis. Hypokalemia is also common secondary to renal K wasting, and muscle weakness is a common complaint. Occasionally, an incomplete form of RTA-1 may occur. Common findings include NG acidosis and alkaline urine with hypocitraturia. However, patients with this incomplete form of RTA-1 are able to maintain serum bicarbonate levels unless stressed.
Type 2 RTA results from proximal tubule HCO3 wasting. Because distal acid excretion is normal in these patients, there is a lower limit for the possible bicarbonate concentration - usually 12-20 meq/L, and alkali therapy results in HCO3 wasting. Urine pH in these cases can be variable. In the setting of an alkali load, the urine ph is elevated but under basal conditions, the urine pH is low. Mild hypokalemia is common.
Type 4 RTA results from decreased aldosterone action either due to reduced hormone level or functional resistance. Acidosis is often mild, with serum bicarbonate rarely decreases below 16-18 meq/L and may be in the normal range. Urine pH commonly falls below 5.5, and hyperkalemia is the most prominent abnormality (and may be the only abnormality noted.) The etiologies of RTA are summarized in Table 3.
The history is an essential part of initial evaluation, though oftentimes, it is not available or simply unreliable. Histories from relatives and prehospital caregivers are important. Old patient records as well as details of recent hospitalization should be thoroughly reviewed.
First, measure arterial pH, PCO2, and serum bicarbonate concentration.
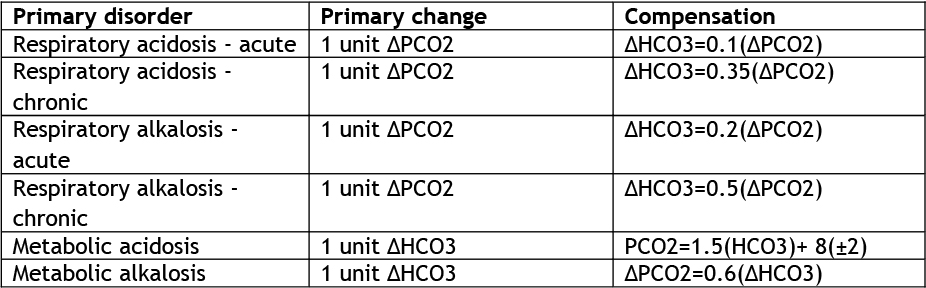
Where a low bicarbonate is present, the anion gap should be checked to determine if this is a gap, non-gap or mixed disorder. The PCO2 will help determine whether there is a superimposed respiratory acidosis or alkalosis. This can be ascertained by calculating whether the degree of respiratory compensation is appropriate (Table 4).
Serum AG=serum Na - serum (Cl +HCO3). The anion gap is a construct that does not truly exist (electroneutrality is always maintained) and represents the difference between the commonly measured anion (Na) and cations (HCO3 and Cl). Thus, the anion gap can change either due to an increase in unmeasured anions or a change in the relative amounts of chloride and bicarbonate. The measured rather than the corrected serum sodium is used for calculations. The AG is comprised primarily of negative charges on serum proteins, primarily albumin. The normal serum AG ranges from 8 to 12 meq/L, but varies between different laboratories. It is important to note that changes in the serum albumin concentration affect the AG and so the calculated AG should be corrected for the albumin concentration. The AG typically decreases by 2.5 meq/L for every 1 g/dl reduction in serum albumin below 4 g/dl.
In complicated mixed acid-base disorders, metabolic acidosis may be less obvious. It is important to establish the primary acid-base disorder first, then check the compensatory response to assess for superimposed acid-base disorders (See Table 4).
To further assess metabolic acidosis, determine the delta AG (ΔAG). ΔAG=AG(measured)-AG(normal). If ΔAG +serum HCO3 <20, then there is NG metabolic acidosis in addition to AG metabolic acidosis. If the delta gap plus serum HCO3 > 28 then there is a metabolic alkalosis as well.
If an AG metabolic acidosis is established, additional tests to determine the causes should be ordered based on the history and physical findings.
The diagnosis of D-lactic acidosis should be entertained in patients with short bowel syndrome. These patients present with AG acidosis, negative ketones, and normal serum lactate level by routine lactate testing. The diagnosis is likely if the acidosis worsens with oral intake. It requires special testing for D-lactate.
Since alcohol delays the metabolism of methanol and ethylene glycol, AG acidosis may not be present in patients co-ingesting significant amount of alcohol. In such cases, elevated serum osmolar gap may be helpful in establishing the diagnosis. The urinalysis may provide important clue to the presence of ethylene glycol. Calcium oxalate crystals are common in the urine (though not specific) if sufficient ethylene glycol has been ingested. The definitive test to diagnose toxic ingestion is to measure the levels of the toxin in the serum. However, the test may not be readily available when there is a need for urgent decision-making.
For a NG metabolic acidosis, the urinary AG is often used to distinguish between renal and extra-renal HCO3 losses. Urinary AG= urine (Na +K -Cl). The primary unmeasured anion is ammonium and in the setting of a metabolic acidosis of non-renal origin, there should be a marked increase in ammonium excretion which is excreted with chloride and thus leads to a negative urinary anion gap. A positive urinary anion gap suggests that there is inadequate ammonium production and suggests the presence of a distal RTA. However, an exception to this would be when there is a freely filtered anion that is causing the acidosis (e.g., hippurate or ketoacids) as these will be excreted in the urine with ammonium and thus the gap may be positive despite the presence of a large amount of acid excretion.
To distinguish between different types of RTA, both urine pH and serum K are helpful. Alkaline urine, hypokalemia and hypercalciuria with evidence of kidney stone formation suggest type 1 RTA, whereas relative acidic urine with hyperkalemia suggests type 4 RTA. In addition, the acidosis in type 1 RTA is usually severe, compared to those in type 2 and 4.
Once NG metabolic acidosis is confirmed, additional testing will be based on the suspected diagnoses. Radiological imaging has limited role in the acute management of metabolic acidosis, except in rare occasions, imaging may be helpful in establishing tissue/organ ischemia, nephrocalcinosis and other processes that are associated with a particular acid-base disorder.
A pH <7.1 or severe acute acidosis with compromised hemodynamics is considered a medical emergency as there may be significant neurological complications and high risks of cardiac arrhythmia. Treatment with intravenous bicarbonate is warranted (Class IIa recommendation). The initial goal is to raise serum pH to 7.15, or serum HCO3 to 15. The initial dose will be based on the HCO3 deficit. Typically 50% of the deficit is given as an iv bolus, the rest is given over 6-12 hours. Occasionally, hemodialysis may be needed for bicarbonate repletion particularly in the setting of volume overload with hemodynamic compromise. Bicarbonate therapy may not be needed. In ketoacidosis (diabetic or alcoholic) since organic anions can be converted to HCO3 rapidly after administration of insulin (DKA) or glucose (AKA). Thus exogenous bicarbonate should be administered with caution in keto acidosis and lactic acidosis to prevent overshoot metabolic alkalosis.
Calculation of bicarbonate deficit. Terms: VOD=volume of distribution in liters; BW=body weight in Kg; HCO3 deficit=VOD for HCO3 x HCO3 deficit per liter. HCO3 VOD varies per serum HCO3 levels. In severe deficiency, the VOD increases significantly due to contributions from intracellular space and bone. Easy rule of thumb: if serum HCO3 >10 meq/L => VOD =0.5 x BW; if serum HCO3 between 5-10 meq/L => VOD =0.75 x BW; if serum HCO3 <5 meq/L => VOD=1 x BW. HCO3 deficit per liter =target HCO3 level - initial serum HCO3 level.
Intravenous NaHCO3 comes as 8.4% solution. To administer an isotonic solution, three amps (150ml) of NaHCO3 are mixed with 1 liter of D5W to get ~150 meq/L of NaHCO3. Oral NaHCO3: one tablet (650 mg) provides ~7.7 meq of HCO3.
The use of bicarbonate therapy for a metabolic acidosis is controversial because of the lack of demonstrated benefits as well as a number of potential complications including: 1) CO2 generation, leading to worsening intracellular acidosis; 2) reduction of ionized serum calcium, as Ca and H+ compete for albumin binding. Rapid increases of pH may lead to more Ca binding to albumin and reduction of ionized Ca; and 3) volume overload.
The use of bicarbonate in cases of organic acidosis (e.g., ketoacidosis and L-lactic acidosis) is also controversial. In patients with severe acidosis (pH<7.1) and compromised hemodynamics, bicarbonate therapy is warranted (class IIa recommendation), since severe acidemia may lead to continued tissue hypoperfusion from reduced cardiac function and impaired oxygen delivery. However, studies thus far fail to show any benefit in hemodynamic improvement or mortality.
For lactic acidosis, it is most important to correct the underlying abnormalities, such as restoration of tissue perfusion and treatment of underlying malignancies.
For diabetic ketoacidosis, insulin therapy needs to be started immediately. At the same time, aggressive fluid and electrolyte management should be instituted.
For D-lactic acidosis, mild cases often do not require treatment except restriction of carbohydrate intake. In more severe cases or patients with symptoms, treatment typically involves administration of NaHCO3 (PO or iv) and oral antimicrobials including Metronidazole or Vancomycin.
For methanol or ethylene glycol intoxication, the serum toxin level may not be readily available at the time of clinical decision-making. Treatment should not be delayed, as permanent end-organ damage may occur if left untreated. Patients should be monitored closely in an ICU setting and managed accordingly for airway, breathing and circulation.
The first step of therapy is to block the metabolism of these parent alcohols. An ethanol drip (iv, 5%-10% solution in D5W, and target serum alcohol concentration of 100 mg/dl) has been used with success. However, CNS sedation, difficulty in dosing and other complications have limited its use. Fomepizole was approved in 1997 as an antidote for methanol and ethylene glycol intoxications. Its binding affinity for alcohol dehydrogenase is 8,000 times greater compared to alcohol and has been proven to be highly effective with minimal complications. It should be started immediately in cases of suspected ingestions.
The typical regimen includes initial loading of 15 mg/kg iv bolus, then 10 mg/kg (increase to 15 mg/kg after 48 hours) iv bolus every 12 hours, until the serum level of parent alcohol is undetectable or below 20 mg/dl and patient becomes asymptomatic with normal pH. The Fomepizole dosing regimen needs to be adjusted in patients receiving dialysis.
After the initiation of therapy with either ethanol or Fomepizole, hemodialysis may be needed to facilitate the removal of the parent alcohol. Hemodialysis should be considered in cases of renal failure, significant or worsening metabolic acidosis, or measured parent alcohol level >50 mg/dl. Of note, thiamine and pyridoxine have been used in ethylene glycol intoxication, as they are involved in alternative elimination pathways of glyoxylate. To date, there is no data to prove their efficacy.
Since urinary excretion of salicylate can be significantly increased in alkaline urine, it is a common practice to induce alkaline diuresis. NaHCO3 iv (150 mmol mixed in 1 liter of D5W) is used to raise urine pH to 7.5 or above. Typically, a rate of 0.5 mmol/kg/hr is initiated and titrated to the target urine pH. Importantly, KCl (usually starts at 40 meq iv) needs to be added to the regimen since body potassium depletion is invariably present and may be masked by acidosis.
It is important to monitor serum potassium and magnesium and replete aggressively. Hemodialysis is effective in removing salicylate and is indicated 1) in patients with serum salicylate level >90 mg/dl (regardless of renal function), or 2) in patients with reduced renal function and serum salicylate level >75 mg/dl, or 3) in those with severe metabolic acidosis (pH<7.1), or 4) in those with severe or progressive clinical decompensation.
For Toluene toxicity, treatment is usually supportive, i.e., maintain hydration, replete potassium and other electrolytes, and use bicarbonate therapy in severe cases (pH<7.2). Recovery is usually rapid. Since Toluene is lipophilic and stored in body fat, dialysis is usually not effective.
In cases of RTA-1, alkali therapy with NaHCO3 is indicated, to correct acidosis and maintain bone health. Bicarbonate therapy also effectively repletes intravascular volume (volume depletion is common in RTA-1). K supplementation is rarely needed. By further raising the urine pH, bicarbonate therapy may increase the risk of calcium phosphate stones in patients with RTA. In RTA-2, alkali therapy is generally not indicated except in pediatric patients, where it has been suggested that RTA-2 is associated with poor growth. If alkali therapy is used, expect a higher than usual dose due to high renal wasting. K wasting will become significant with alkali therapy, and supplementation is often required.
Of note, there is anecdotal success with thiazide diuretic in RTA-2. It causes slight intravascular volume depletion and therefore, stimulates HCO3 reabsorption. In RTA-4, dietary K restriction and diuretic for volume control are the mainstay of therapy. Fludrocortisone (at a dose of 0.05mg to 0.2mg PO daily) is commonly used in patients with primary adrenal insufficiency, though its use in secondary hypoaldosteronism is limited by hypertension and volume overload.
Patients with metabolic acidosis often present with nonspecific symptoms, including headache, chest pain, palpitation, shortness of breath, nausea, vomiting, muscle weakness, and bone pain. In some patients, there may be rapid deep breathing, anxiety, and change in mental status. Severe acidosis can lead to seizure, coma, cardiac arrhythmia and arrest. If metabolic acidosis is recognized and treated promptly, patients may not experience any long-term complications.
For patients with metabolic acidosis from methanol or ethylene glycol, central nervous system (CNS) sedation is a common manifestation. As mentioned earlier, the main toxicities come from metabolites of these parent alcohols. The final metabolite of methanol (formic acid) is highly toxic to the retina, and can lead to permanent blindness. The final metabolites of ethylene glycol target the kidney primarily, and lead to acute tubular injury and tubular obstruction from oxalate crystallization.
In the early stage of methanol intoxication, patients may be relatively asymptomatic, but worsening CNS sedation and cardiopulmonary decompensation may develop soon after. Survivors have a high incidence of permanent blindness. Gastrointestinal symptoms are also common.
There are several stages of ethylene glycol intoxication. Stage 1 occurs up to 12 hours after ingestion. Patients present with acute alcohol-like intoxication. CNS sedation of varying degree is common, and in severe cases, arrhythmia may occur as a result of decreased serum ionized calcium. Stage 2 occurs from 12 to 24 hours after ingestion. Patients experience cardiopulmonary symptoms, including tachycardia, tachypnea and in severe cases, shock. Stage 3 is usually the late stage, occurs 24 hours after the ingestion. Patients develop acute kidney injury, commonly, oligoanuric renal failure.
Patients with significant salicylate overdose may present with coma, with or without hyperventilation. Prognosis is related to the serum salicylate level, age, comorbid illnesses and degree of clinical decompensation.
Patients with RTA-1 typically experience progressive bone resorption from persistent acidosis if left untreated. They are also more likely to develop kidney stone disease due to high urinary calcium excretion, high urine pH and low urinary citrate excretion.


No comments:
Post a Comment